主题:【原创】搭积木 -- 喜欢喝冰茶
家园 【原创】搭积木 搭积木也算科学版
 ,应该放到宝宝版里才对
,应该放到宝宝版里才对 。您先别急,俺是标题党,且听俺慢慢道来
。您先别急,俺是标题党,且听俺慢慢道来 。
。小时候大家都玩儿过积木,后来高级点儿的就改塑胶了,再大点儿就上螺丝刀装模型了,不知道现在的小孩子们都玩儿什么高级玩具了。不过俺要讲得要比这玩艺儿复杂点儿,是给大人玩儿的。如果你有很多块儿积木,但是只有二十种,那怎么搭才能搭成一个稳定的东西呢?
呵呵,有河友已经猜出俺要唠叨什么了
。没错儿,我们每天健康的生活着,就是因为我们的细胞们正常的工作着,或者说生物分子们稳定地行使着它们的功能。这些家伙们主要是两类,脱氧核糖核酸(DNA)/核糖核酸(RNA)和蛋白质(protein)。DNA/RNA主要担负着传递遗传信息的重任,而每天的吃穿用度就靠蛋白质们扛着了。蛋白质的最基本单位叫氨基酸(amino acid),总共有二十种,班主已经在他/她的文章里介绍过了,这里俺就说说这20种积木怎么构建成一个功能的实体。学生物的都知道结构与功能的关系(这也是大白话,没有结构怎么会有功能,功能肯定依赖于结构
 ),所以掌握蛋白质的结构一直是一个很有挑战性的话题。那么蛋白质的结构是怎么样的呢?既然蛋白质是由20种氨基酸组成的,所以这些氨基酸序列就构成了蛋白质最简单的结构----一级结构(primary structure),就像一个简单的字符串儿(幸好只有20个氨基酸,而英文有26个字母)。但是这么个一维序列显然不能描述蛋白质的空间结构,于是二级结构(secondary structure)的概念被引入。在蛋白三维结构中,有很多相同结构特性的单位,像alpha-helix,beta-sheet,(图里面的螺旋和那些带箭头的带子)
),所以掌握蛋白质的结构一直是一个很有挑战性的话题。那么蛋白质的结构是怎么样的呢?既然蛋白质是由20种氨基酸组成的,所以这些氨基酸序列就构成了蛋白质最简单的结构----一级结构(primary structure),就像一个简单的字符串儿(幸好只有20个氨基酸,而英文有26个字母)。但是这么个一维序列显然不能描述蛋白质的空间结构,于是二级结构(secondary structure)的概念被引入。在蛋白三维结构中,有很多相同结构特性的单位,像alpha-helix,beta-sheet,(图里面的螺旋和那些带箭头的带子)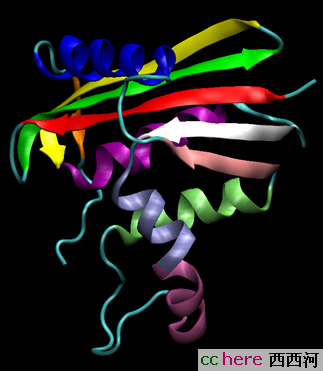
这些都是典型的二级结构,而一条氨基酸链的空间结构则被称为三级结构(tertiary structure)。但是很多protein包含不止一条的氨基酸链,所以四极结构(Quaternary structure)的概念被引入来表达这种复杂的多体结构。
现在我们知道Protein有四极结构。我们身体里有成千上万的蛋白,那人们怎么知道这些东西都长什么样子呢?最先发展起来的实验技术要感谢伦琴同学的卓越工作,他老婆的手照片在科学史上的地位赶得上蒙娜丽莎的微笑在卢浮宫的地位了
。没错儿,最常用也是比较成熟的实验技术就是X-ray晶体技术,这种技术通过在特定的外界条件(一般都是比较极端的条件)下,使得纯化的高纯度蛋白质结成晶体,然后分析X-ray衍射结果,从而推出蛋白质的空间结构。另外一种常用技术则是核磁共振技术(Nuclear magnetic resonance spectroscopy, NMR),可能有河友不太熟悉这个东西,但是它的姊妹技术----磁共振成像技术(Magnetic resonance imaging, MRI)已经广泛的应用于生物医学领域,现在有点规模的医院一般都有。核磁共振技术的优点主要在于,不像X-ray晶体技术,它可以直接测量水溶液中蛋白的结构。这一点很重要,因为水是生命的母亲,有水就能孕育生命,所以那些太空探索者们如果在某个星球发现有水的痕迹,一般都跟发了财似的兴奋。而X-ray的晶体一般都不是在生理环境下长成的(多稀罕啊,要是生理环境下都结晶,那我们可就麻烦大了),那么一个重要的问题就是谁能保证正常生理环境下这些蛋白还长这样子?所以X-ray的研究人员必须参考其它生物物理/生物化学的结果,来证明结晶结构是合理的。那为什么核磁共振技术不能替代X-ray方法呢?因为根据现在的数据处理能力,稍微大些的蛋白质的NMR结果 --- 核磁共振谱,很难解出来,也就是说,大点儿的protein靠NMR解不出结构来,所以X-ray仍然坐着老大的交椅。现在大约有4万多个蛋白结构通过实验方法得到,但是有些是一些片断或者同一蛋白的突变体结构,也就是冗余(redundent)结构,去掉它们大概也只有两三万个可以用的。这么多结构当然需要管理,数据库是最好的方式,所以公用数据库Protein Data Bank(PDB)诞生了。每个蛋白结构被存为一个简单的文本文件,除了坐标,里面还包括序列,试验参数,等等。我们身体里的蛋白质的数量远远大于4万个,所以实验者们的路还很长。但是,晶体学本身还不是一门完全成熟的学科,Protein结晶的条件更多地依赖于经验,所以研究人员们面临一个巨大的挑战就是,基于当前的实验技术,不是所有的蛋白质(例如大部分膜蛋白)都能得到结构的。那可怎么办?
还好,由于数学理论和计算技术的发展,生物计算得以广泛的使用,其中有一门技术称为蛋白质结构预测(protein structure prediction),让人们看到了一线曙光。蛋白质结构预测的基本思想就是从一极序列,也就是字符串开始,利用各种数学方法和计算理论,推测出(或者说猜出也行)蛋白质的空间结构。主要有三种办法。第一种称为Homology Modeling,同源模型(是这么叫的吗?)。那么什么是生物的同源性呢?简单的说,人的肌肉蛋白可以让我们运动,而兔子也有对应的蛋白让它运动,所以这两种蛋白可以称为同源蛋白,也就是说在进化图上,很有可能是从同一个祖先蛋白而来。一般来讲,同源蛋白的一极序列,也就是字符串比较像,Homology modeling的思想就是假设同源蛋白的结构非常相似(很多实验证据支持这一假设),通过“对齐”(术语叫Alignment)几个同源蛋白的一极序列,只要其中有一个的结构是已知的(这个一般叫模版),那么就可以推测出其他蛋白的结构。有意思得是所谓的"对齐"并不一定要求相同的氨基酸,相似的对齐就行。这儿所说的相似指得是氨基酸的物理化学性质相似。虽然有20种氨基酸,但是就物化性质来划分只有5类,所以相似性还是很有意义的。在实际操作时,如果序列的相似性similarity(不是一致性identity哈)不小于35%,这种方法基本上还是比较可信的。但如果小于35%如何?于是引入第二种方法,Threading/Fold recognition(这个术语的中文不知道叫什么
 ),简单的说就是因为两条序列总体不是很像,但是局部还是比较近似,所以“拟和”局部结构,例如蛋白质的二级结构,剩下的部分只好"猜"了。当然所谓的"猜"也不是瞎猜,可以结合一些其他的计算办法,例如能量最小化,推测出这部分结构。如果真的运气不好,实在找不到模板,那就只好"抓阄"了,也就是第三种方法,ab initio,最后的办法。这种方法的基本操作是这样的,初始输入是一维的蛋白质序列,每个氨基酸用一个点表示,之间用线相连,这些点可以移动,然后定义一些能量函数,例如我们都知道同性电荷相斥,所以两个都带正电的不直接相连的氨基酸不太愿意呆在一起,那么就可以定义如果两个同性电荷靠在一起,就设成一个正数,而异性电荷则设成负值。每次随机让这个链儿动动,所以新状态的能量函数就可能和老的不一样。现在人们认为蛋白质的稳定结构应该是一种能量最小的结构,因而如果能量减少,我们就接受这种新的样子(构象,Conformation)。但是如果增加是不是一定要舍弃呢?不行,简单的讲,是因为会掉进沟里出不来。看下面这张图。
),简单的说就是因为两条序列总体不是很像,但是局部还是比较近似,所以“拟和”局部结构,例如蛋白质的二级结构,剩下的部分只好"猜"了。当然所谓的"猜"也不是瞎猜,可以结合一些其他的计算办法,例如能量最小化,推测出这部分结构。如果真的运气不好,实在找不到模板,那就只好"抓阄"了,也就是第三种方法,ab initio,最后的办法。这种方法的基本操作是这样的,初始输入是一维的蛋白质序列,每个氨基酸用一个点表示,之间用线相连,这些点可以移动,然后定义一些能量函数,例如我们都知道同性电荷相斥,所以两个都带正电的不直接相连的氨基酸不太愿意呆在一起,那么就可以定义如果两个同性电荷靠在一起,就设成一个正数,而异性电荷则设成负值。每次随机让这个链儿动动,所以新状态的能量函数就可能和老的不一样。现在人们认为蛋白质的稳定结构应该是一种能量最小的结构,因而如果能量减少,我们就接受这种新的样子(构象,Conformation)。但是如果增加是不是一定要舍弃呢?不行,简单的讲,是因为会掉进沟里出不来。看下面这张图。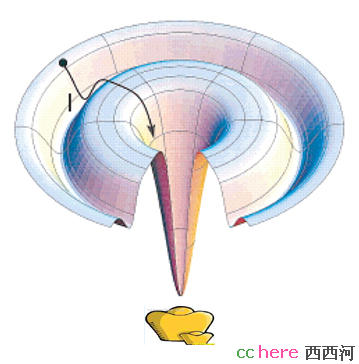
假定一个人去寻宝,宝藏藏在最低的谷底,但是峭壁很滑,如果寻宝者一味的向低处走,他就很有可能掉进一个山谷(例如图中的I点),但却不是最深的谷底,而被陷在里面。在ab initio方法中,寻求更低能量的蛋白质新构象,相当于寻宝者向低处走,所以一味的降低能量,蛋白质有可能就陷在非最小的能量谷底(局部最小值,local minimum)。既然我们同意蛋白质的稳定结构应该是能量最低的,所以正确的做法应该是让输出的蛋白质新构象的能量最低,也就是全局最小值(global minimum)。那么如何解决这个问题呢?就是当能量增加的时候,不是简单的舍弃这种构象,而是引入一个概率能量因子,它给新老状态的能量相关,然后随即产生一个概率,比较它们以决定取舍,从而降低陷在local minimum的概率。循环这个操作直到能量达到期望为止。所以简单来说,ab initio过程就是数学上求解函数的Global Minimum,只不过这个函数没人知道。
在实际操作中,经常是多种方法合用,一起找到最好的可能结构。可能细心的河友会问,唠叨了这么多,那我怎么知道你算的对不对啊。A good question, 事实上,这个问题是所有生物计算领域的一个最基本也是最常问的问题。很不幸,没有直接的证据能证明,你想啊,要是做实验的都能在实验中看到或者做到,那就不会有生物计算这个领域了,所以不可能有来自于试验的直接证据,但是间接证据却有好多,有些计算技术也越来越被人们所认可。但是就蛋白结构预测来讲,三种技术中比较可信的还是第一种,后两种相对来讲不太可信。但是前途是光明滴,虽然道路是曲折滴。从两年一届的擂台赛CASP(Critical Assessment of techniques for protein Structure Prediction)的记录中可以看出,相对于以前,成功率还是在提高。但离最终的目标还有距离,所以人们还要不停的试呀,搭呀,直到用积木搭出一个大家都满意的东西来为止。
元宝推荐:游识猷,海天,爱莲,
本帖一共被 1 帖 引用 (帖内工具实现)- 复 【原创】搭积木
家园 今天老是花出宝来 原来冰茶兄是科学人啊。
恭喜:你意外获得【西西河通宝】一枚
鲜花已经成功送出。
此次送花为【有效送花赞扬,涨乐善、声望】
- 复 【原创】搭积木
家园 问个外行问题 说到aa序列时候,常常用domain这个术语
domain到底是如何来发现的。现在可能更多的是序列比对,但是最早的时候是怎么得来的?是单纯根据序列,还是用一些理化手段处理蛋白质,发现一个domain里面的aa能有比较类似的结果?
- 复 问个外行问题
家园 Domain最早还是个structure的概念 那些比较稳定的相似结构就称作domian。所以早期的domain是从crystal structure上发现的。后来又发现一些domain在不同的protein里具有相似的功能, 比如DNA-binding, 于是就有了functional domain的概念。然后又发现那些具有相似功能的domain在AA序列上有一定的规律,这才有搞计算机的人们在上面做文章。
- 复 问个外行问题
家园 我想最先还是先根据实验室生化性质判定出来的吧 比如zinc finger,肯定是先有物理化学性质,再推断类似结构成为一个模式的。
- 复 问个外行问题
家园 Domain这东西哈 基于俺这二把刀的理解,一般指结构上的。Wiki上有很多解释,个人的感觉吧,就是一"特有型"的结构单位
,介于二级和三级之间的一种东西。像EF构型的钙粒子Binding区,就是两三条alpha-helix组成的,有点儿像用手的虎口夹个球儿似的。最早估计应该是结构方面的实验结果,但是地球人都知道,一级序列决定高级结构,只不过怎么个决定法,咱们就不知道了 。所以干这行的最高理想就是给我一些不同形状的砖头,我能搭一个稳定且有用处的大楼
。所以干这行的最高理想就是给我一些不同形状的砖头,我能搭一个稳定且有用处的大楼 (是不是有点老阿的口气)。
(是不是有点老阿的口气)。
- 复 【原创】搭积木
家园 现在ab initio比较火 最近一期的自然杂志上有篇文章用Rosetta做prediction,结果貌似不错。为此E.Dodson还专门写了篇短文捧场。