主题:【原创】这是真的吗?Maxipreps concentration=4163.1ng/ul?! -- 大鹏翔宇
家园 【原创】这是真的吗?Maxipreps concentration=4163.1ng/ul?! 今天犯小人,最近又太累了,就先不填坑了,大鹏还要在地幔里呆上一段时间。
---------------------------------------
现在向河里生物专业的大牛们请教个问题,大家知道俺是个实验室里的新丁,现在正在练习实验技术。
导师给了两个质粒(plasmid),一个叫phRL SV40 DELTA 197,一个叫phRL TK DELTA 238。都是溶在Whatman 3MM paper上(跟咱河里三好MM没关系啊)送来的。我把它们用buffer溶了,transform进E.coli JM109然后inoculate进5ml LB media,做Minipreps提纯质粒以后,做double digest再跑Agarose Electrophoresis检查正常。
上个星期我花了三天用Minipreps提纯的质粒做了个Maxipreps,用的是Promega的Wizard Maxipreps Kit。做好后今天用Nanodrop Spectrophotometer测了浓度。
 外链图片需谨慎,可能会被源头改
外链图片需谨慎,可能会被源头改 外链图片需谨慎,可能会被源头改
外链图片需谨慎,可能会被源头改 外链图片需谨慎,可能会被源头改
外链图片需谨慎,可能会被源头改这是检测结果:
phRL TK DELTA 238
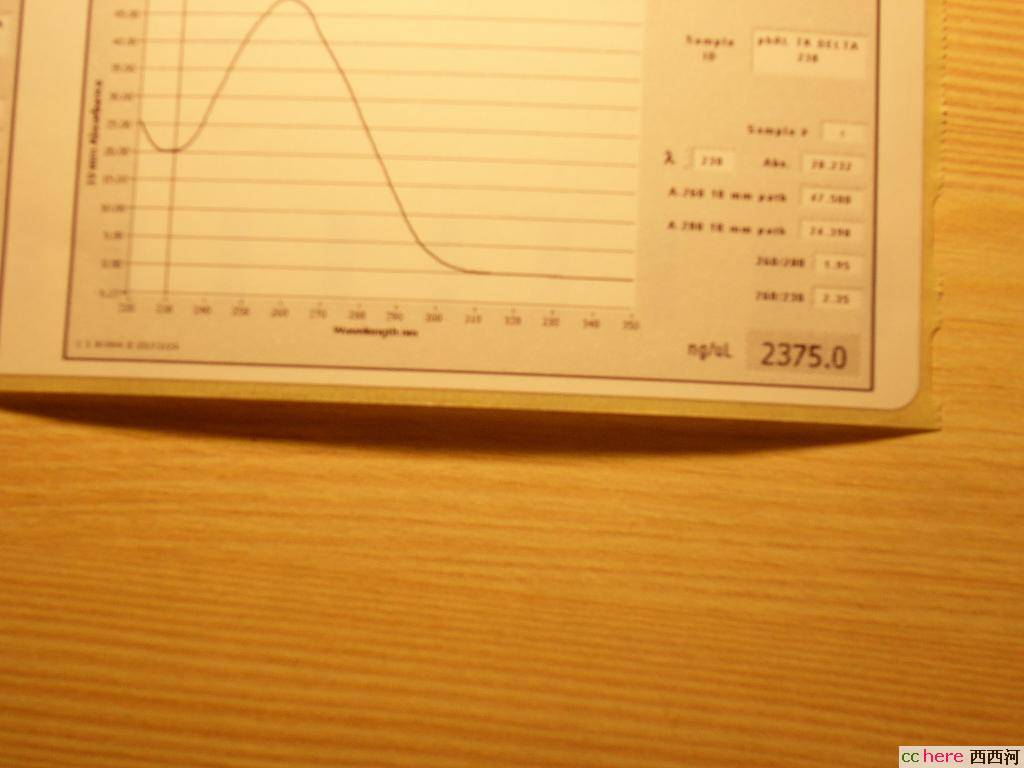
这个还算正常,
phRL SV40 DELTA 197
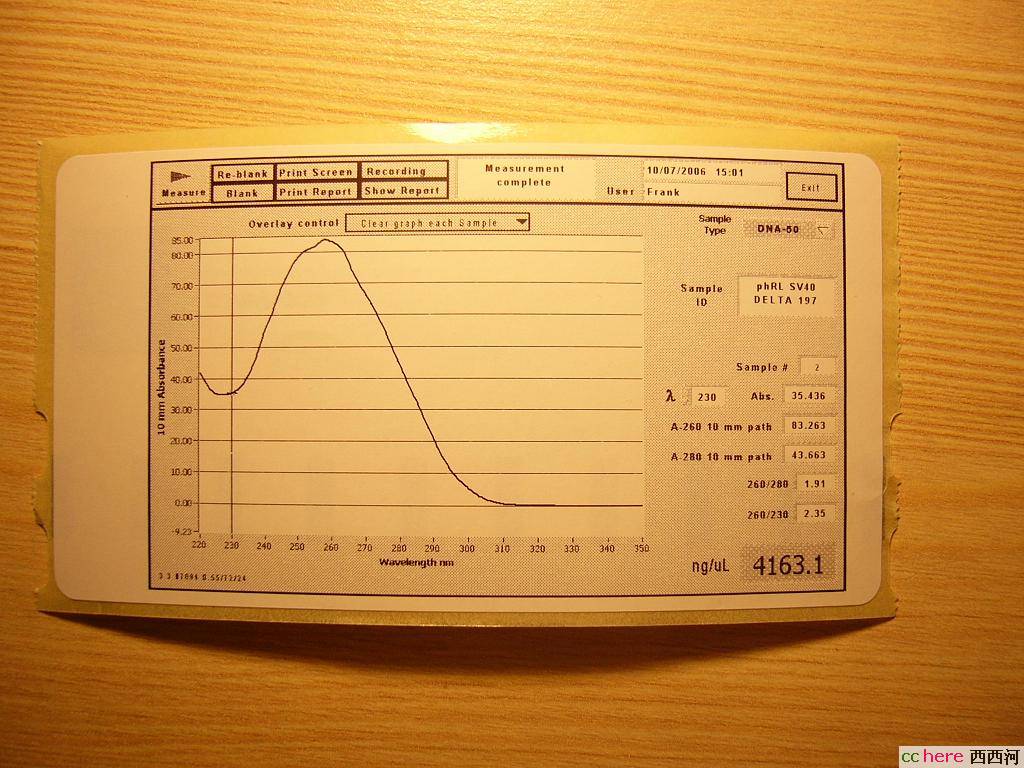
这个浓度让试验室里所有师兄师姐大眼瞪小眼,“This is increditable!”“Unbelievable!”
大鹏有点摸不着头脑了,不是吧?哪里做错了?
印度师姐索尼说了(没错,她就叫Sony):“Usually it is between 2000 and 3000,sometimes gets higher than 3000,but 4163.1,that is too high!”
啊?太高了?会不会有啥问题?
东北师兄过来说:“兄弟啊,要不咱跑个胶看看?”
跑就跑!
胶体电泳结果:
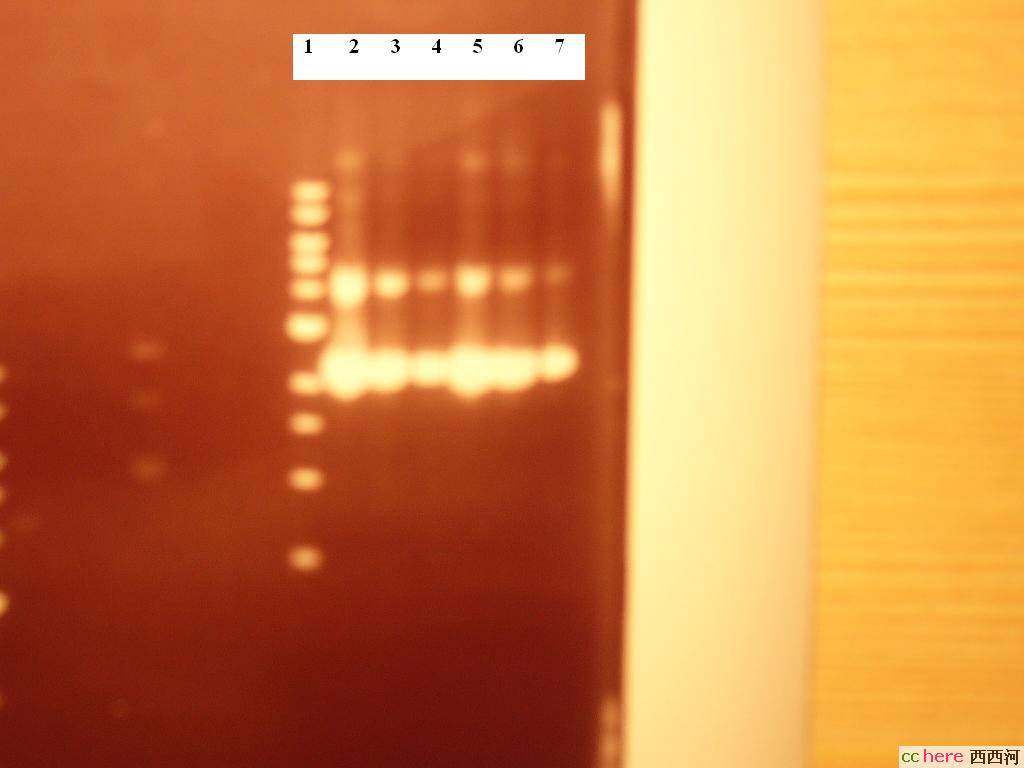
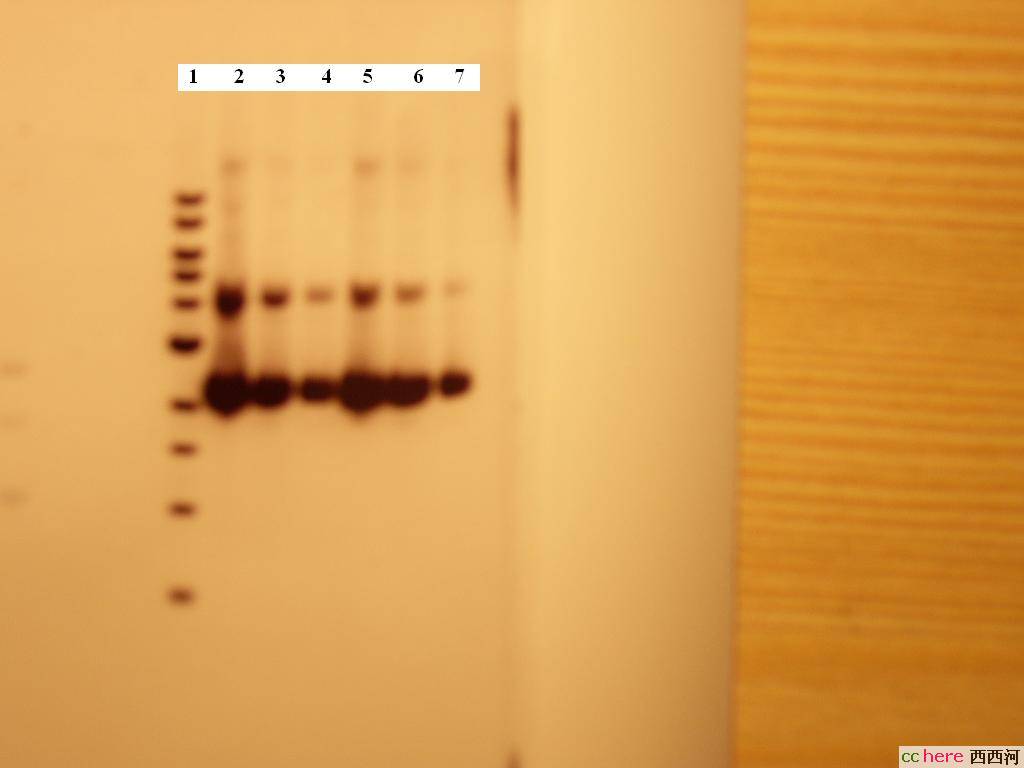
1为DNA 1kb ladder
2-4为phRL SV40 DELTA 197,依次为10×,20×,100×倍稀释的质粒
5-7为phRL TK DELTA 238,依次为10×,20×,100×倍稀释的质粒
根据条带的亮度看来的确是非常浓的质粒溶液,不过Promega给出的Maxipreps浓度也就是2000-3500ng/ul,这个是不是太高了点?
期待高人的解答。
另外,可否请教一下那些专业名词的中文说法,大鹏的专业是用英语学的,爸妈问三年都学了些啥,张了半天嘴愣是说不出来,都是英语啊,真惭愧!
在此一词一花悬赏征名!
另:最近lab要搬家,等腾出时间来要在科技版写写lab里的人和事,暂定题目:
“萨达姆哪里拱”---一个中国学生眼中的英国生物实验室
元宝推荐:海天,家园 应该没问题。 多少毫升菌液?曾用500毫升菌液提得4毫克多质粒,还是20KB左右的。所以和商家的最高提取量没多大关系。不过现在都用Gerard Biotech的midi kit,50ml菌液基本上能提1毫克,最多的一次100ml提了5毫克还多,不过用这个试剂盒得从Promega另外定resin去除内毒素,总的说来很不错的一牌子,优点是价格出奇地低。
看来小兄弟手艺不错,是干这些活的料子,前途光明
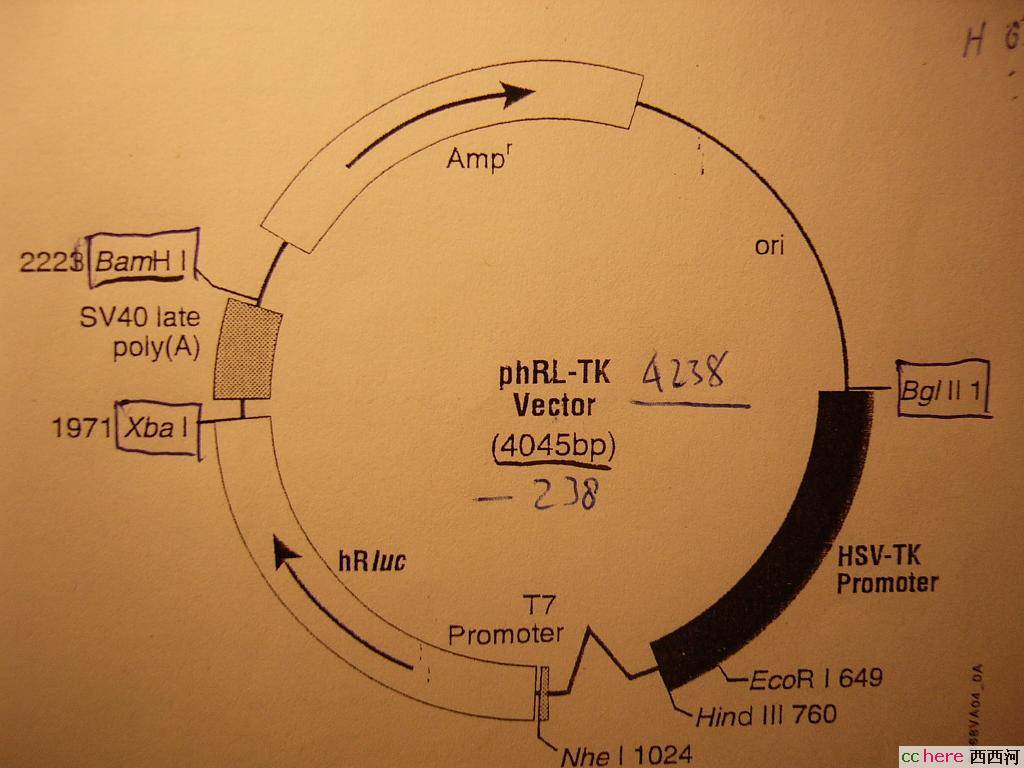
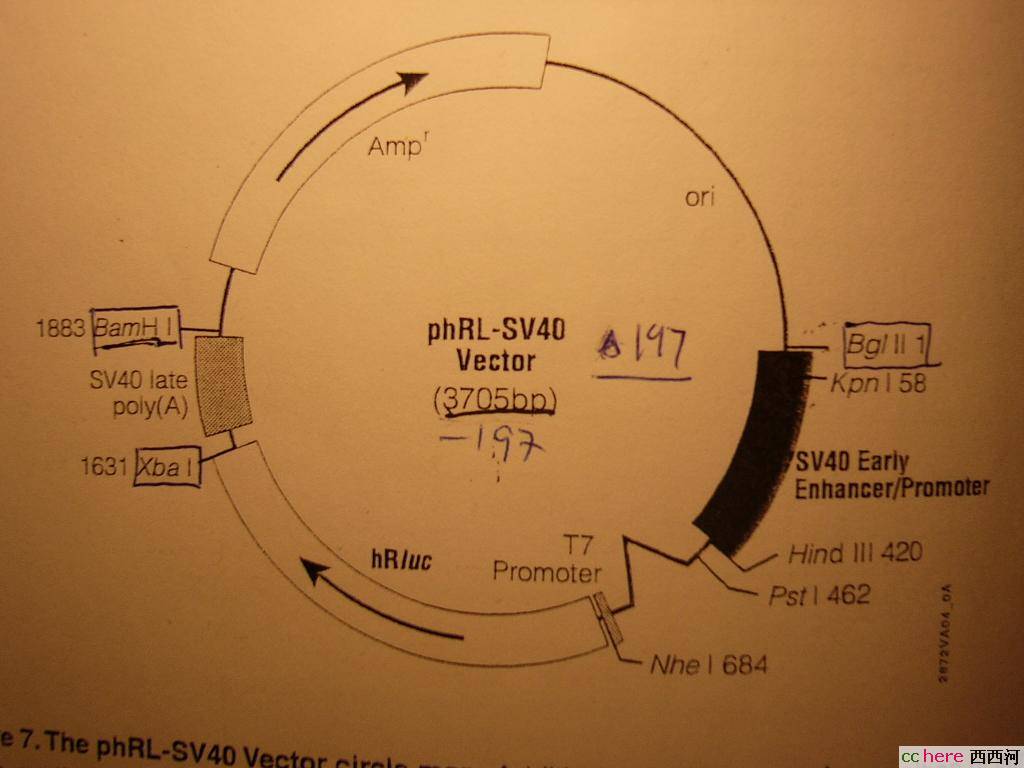
家园 酶切的结果出来了 质粒图:
TK
SV40
电泳图:
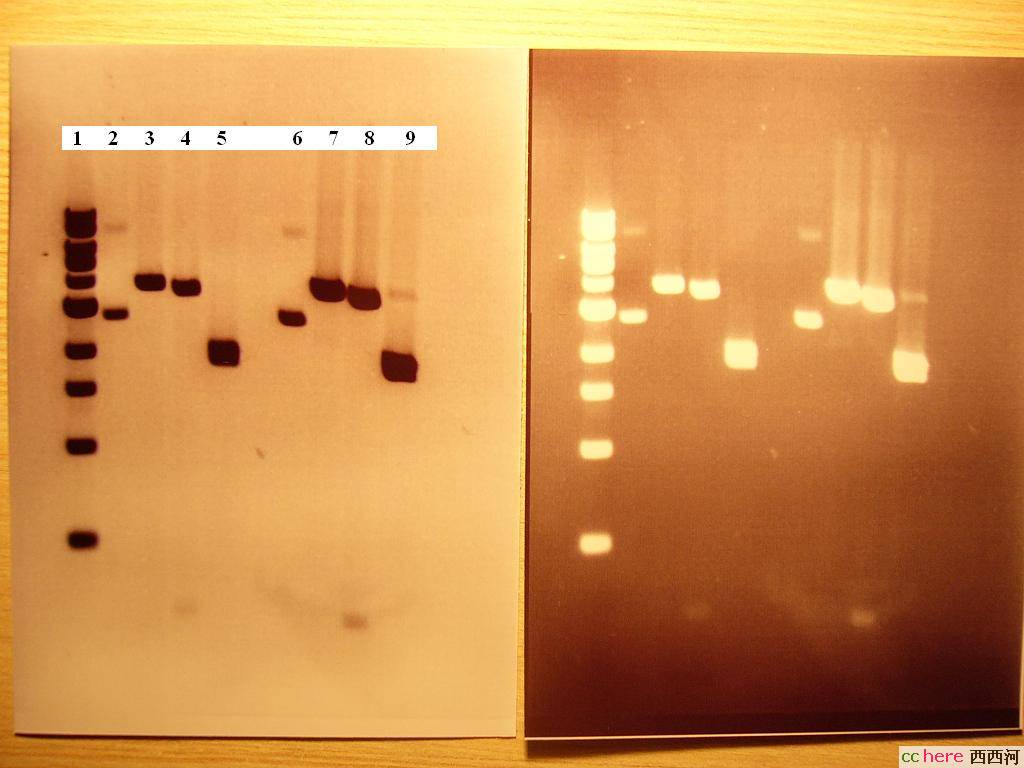
1.DNA 1kb ladder
2.TK(1000×dilution)
3.TK+BamHI
4.TK+BamHI+Xba-I
5.TK+BamHI+BglII
6.SV40(1000×dilution)
7.SV40+BamHI
8.SV40+BamHI+Xba-I
9.SV40+BamHI+BglII
师兄师姐说看上去没问题,还说这个浓度破了实验室的记录了。
这个酶切对吗?
- 复 酶切的结果出来了
家园 Small questions/suggestions Nice figure.
I have several questions/suggestions, though.
1. It would be more informative if you could label the DNA ladder, I tried to guees the size but I could not correlate the sizes.
2. You may want to try to load less sample. It looks to me that you've loaded more than you need - or may be it's just the result of photographying. As a matter of fact, you might be able to distinguish the two bands in lane 5/9 if you chose carefully the amount of sample loaded and the gel concentration.
3. Load the empty lane with loading buffer (prefereably the same ionic strenght as the samples) might help to straighten the running lanes.
4. Lane 9 shows you have not completely digested the sample. The tails in Lane 7-9 may indicate that you have some impurities in your phRL-SV40 vector preparation. This might explain partially why you got the high yield - you could get some E coli genome DNA with your plamid. However, this is just my guessing.
There is a step after you broke the E coli where you need mix that gelly stuff with buffer. You need to mix them gentlely, otherwise, you might break some genome DNA, which will not be separated with centrifugation since it will not precipitate. In another word, it will go with your plasmid sample.
[The above are just my own opinion. We can discuss them if you have different thoughts.]

家园 谢谢指教,有几点解释和疑问 这是一个1kb ladder,不可否认没有分开,其实这个电泳跑得并不好,因为做大胶的设备被打包了(要搬家),只能用小胶但小胶板只能做8个大槽,显然不够,所以做了15个小槽。
样品的确是加多了,所以看起来有点模糊,从上之下依次为:10kb,9kb,8kb,7kb,6kb,5kb+4kb(连在一起,就是紧贴着2上面的那一条),3kb,2kb,1kb,0.5kb。
sample7-9跑歪了,因为跑胶一开始的时候盛胶的塑料盘有点歪。您的方法我下次试试。
您说的不纯导致的浓度偏高可能是对的,是不是需要再分离一次呢?
家园 More question Is your vectors empty or you have already put something in it (1.8 - 2k bps?)? It looks to me that the size of DNA fragments in gel are larger than what it should be based on the plamid map.
您说的不纯导致的浓度偏高可能是对的,是不是需要再分离一次呢?Usually I would not bother to do it if it's a vector. Reason: 1. If you will use it as stock, only plasmid can transform when you want to amplify it; 2. If you want to use it for ligation, you will purify the fragment after the digestion, which will separate them. However, it may consume some of your enzyme, you need take it into account.
家园 我是要用来做transfection的 Is your vectors empty or you have already put something in it (1.8 - 2k bps?)? It looks to me that the size of DNA fragments in gel are larger than what it should be based on the plamid map.对不起,这一段我看得不明白,您是指哪一段呢?

家园 Lane 3/7 Take lane 3 for example: the plasmid is 4045 bp according to the map while the DNA fragment in lane 3 is somewhere around 6k bp. That's why I said you have already put something (a gene) in the vector.
I think you should have since you will use it for transfection. Are you going to use luciferase as a reporter gene?
- 复 酶切的结果出来了
家园 鹏弟啊,看了我一声叹息。 老了!


