主题:【原创】这是真的吗?Maxipreps concentration=4163.1ng/ul?! -- 大鹏翔宇
今天犯小人,最近又太累了,就先不填坑了,大鹏还要在地幔里呆上一段时间。
---------------------------------------
现在向河里生物专业的大牛们请教个问题,大家知道俺是个实验室里的新丁,现在正在练习实验技术。
导师给了两个质粒(plasmid),一个叫phRL SV40 DELTA 197,一个叫phRL TK DELTA 238。都是溶在Whatman 3MM paper上(跟咱河里三好MM没关系啊)送来的。我把它们用buffer溶了,transform进E.coli JM109然后inoculate进5ml LB media,做Minipreps提纯质粒以后,做double digest再跑Agarose Electrophoresis检查正常。
上个星期我花了三天用Minipreps提纯的质粒做了个Maxipreps,用的是Promega的Wizard Maxipreps Kit。做好后今天用Nanodrop Spectrophotometer测了浓度。



这是检测结果:
phRL TK DELTA 238
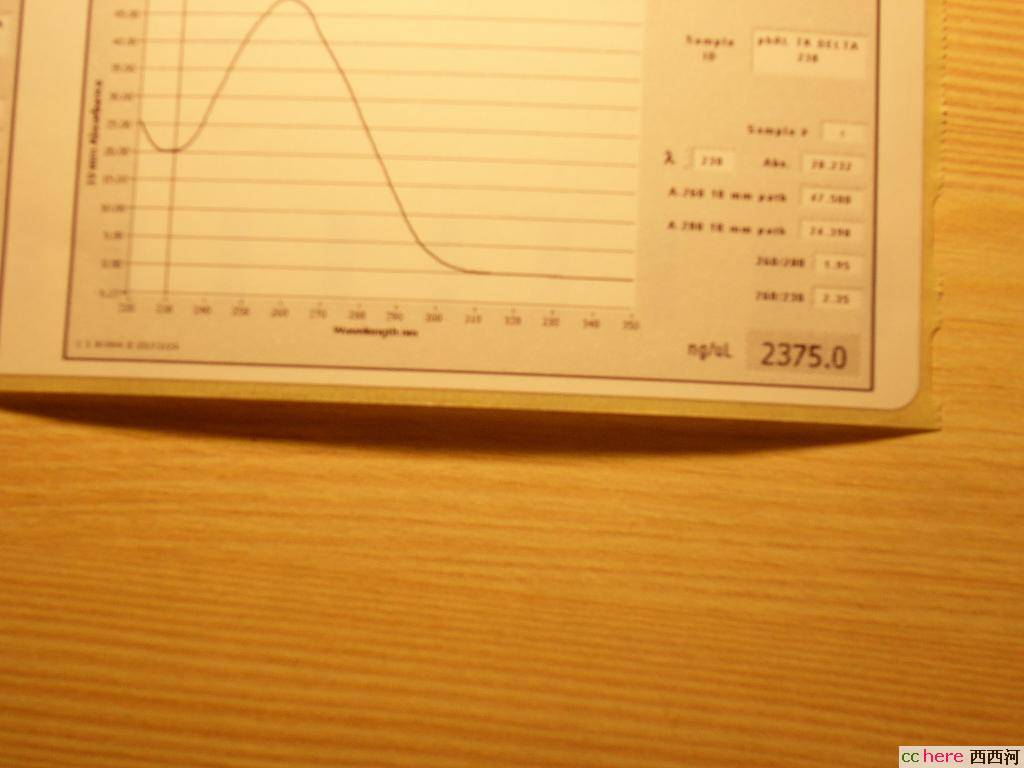
这个还算正常,
phRL SV40 DELTA 197
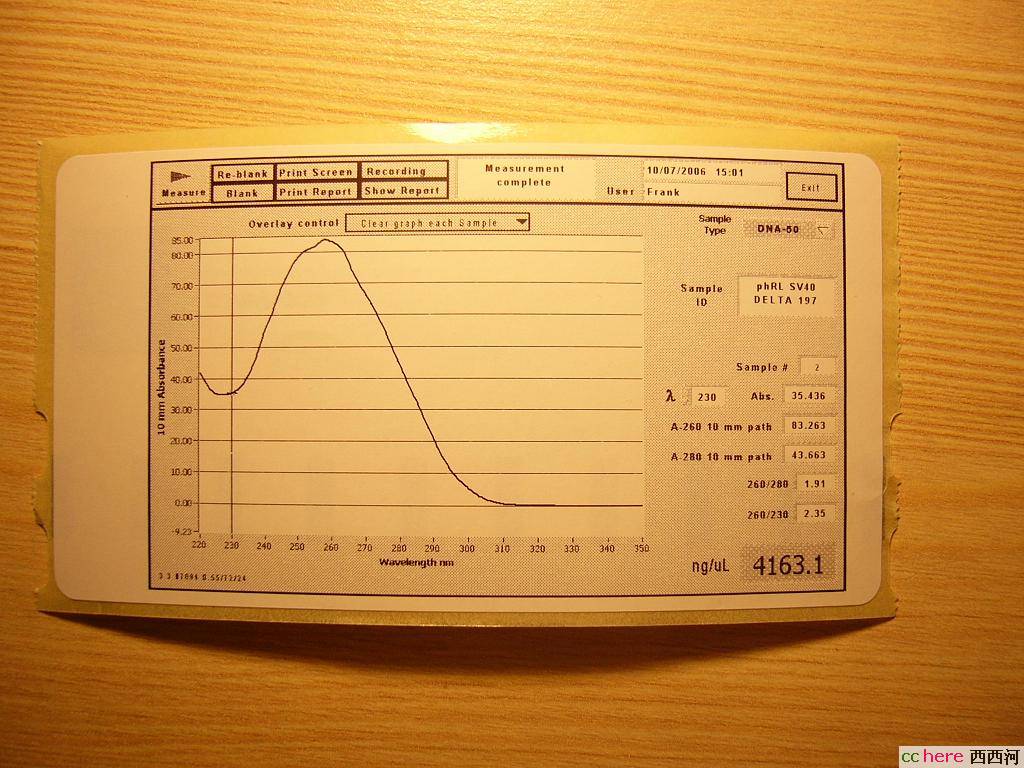
这个浓度让试验室里所有师兄师姐大眼瞪小眼,“This is increditable!”“Unbelievable!”
大鹏有点摸不着头脑了,不是吧?哪里做错了?
印度师姐索尼说了(没错,她就叫Sony):“Usually it is between 2000 and 3000,sometimes gets higher than 3000,but 4163.1,that is too high!”
啊?太高了?会不会有啥问题?
东北师兄过来说:“兄弟啊,要不咱跑个胶看看?”
跑就跑!
胶体电泳结果:
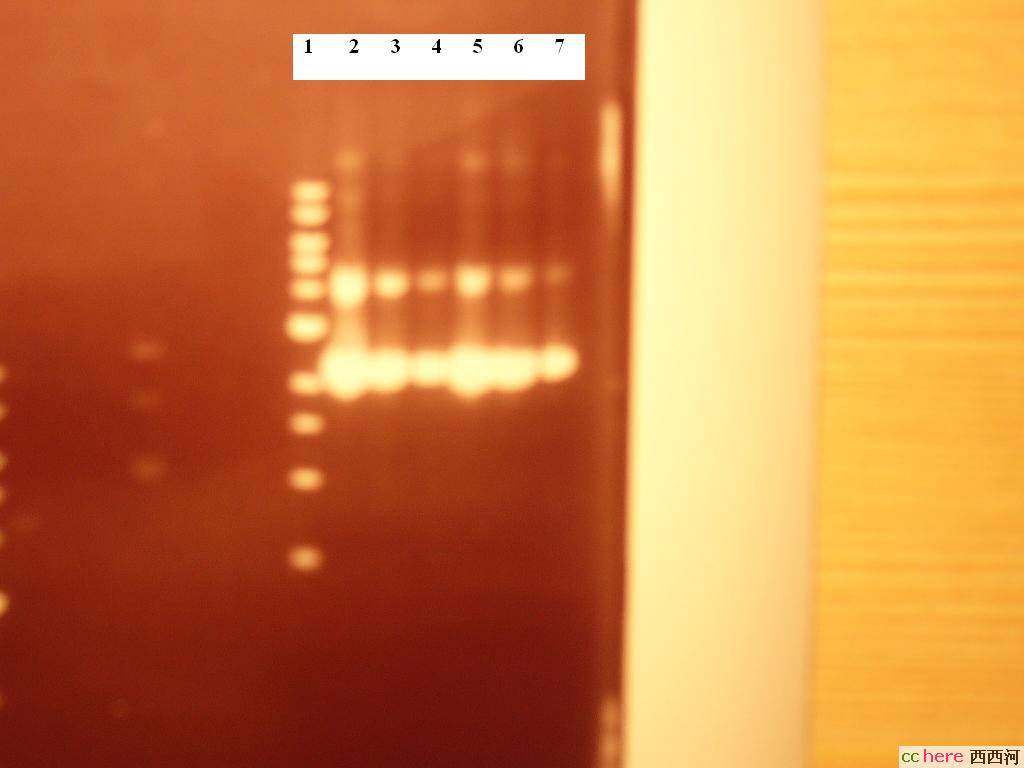
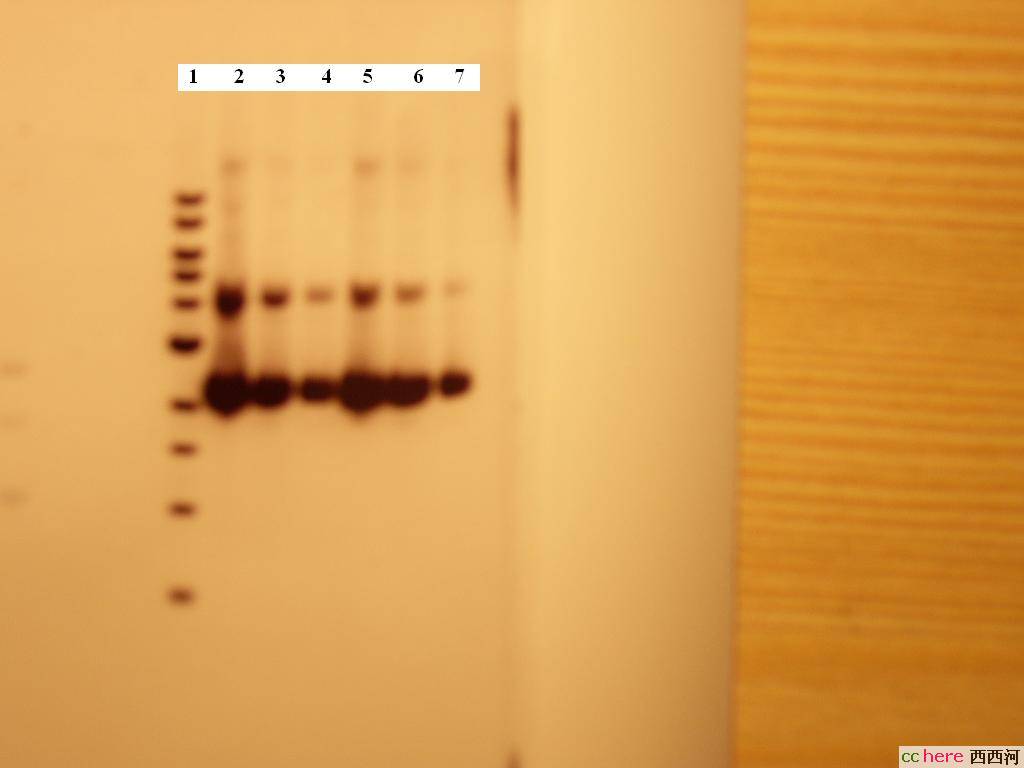
1为DNA 1kb ladder
2-4为phRL SV40 DELTA 197,依次为10×,20×,100×倍稀释的质粒
5-7为phRL TK DELTA 238,依次为10×,20×,100×倍稀释的质粒
根据条带的亮度看来的确是非常浓的质粒溶液,不过Promega给出的Maxipreps浓度也就是2000-3500ng/ul,这个是不是太高了点?
期待高人的解答。
另外,可否请教一下那些专业名词的中文说法,大鹏的专业是用英语学的,爸妈问三年都学了些啥,张了半天嘴愣是说不出来,都是英语啊,真惭愧!
在此一词一花悬赏征名!
另:最近lab要搬家,等腾出时间来要在科技版写写lab里的人和事,暂定题目:
“萨达姆哪里拱”---一个中国学生眼中的英国生物实验室
量多管他呢,说不定你人品爆发呢?查查看这个质粒什么复制子(ori),也许是特别强的。
翻译:
缓冲溶液, 转化, 接种, 双酶切, 琼脂糖电泳, 分光光度计。
我做封闭用的鲑精DNA,浓度是10ug/ul,就像浆糊一般,呵呵。不过实话实说,这样高浓度的DNA不太方便用,太粘了,换我肯定先稀释了再说。
要说maxiprep KIT的产量,是个相当讲人品的地方,QIAGEN的kit,标准产量750ug,我自己的yield,低到1XXug,高到2mg,如果2mg的你愿意只拿500ul水溶,浓度也差不离。
不过,有些问题好像说没太明白。你做maxprep的细菌液是多少?
跑电泳时候好像应该先酶切的,内切酶是什么,可以和酶切图谱多做几个切点的检测,图谱对了就行。
检测浓度的操作有没有问题,测了几次,稳定么?
另外,具体含义理解了,那些单词不知道中文也罢,因为你看文献9层以上用到的是英文。
牛剑麻省斯坦福,大牛真多啊
细菌液浓度没量,因为看上去很浑浊。
酶切没作,因为有四重的安比西林筛选,估计不会有污染。
检测的时候在别人指导下就做了一次,但样品经过充分搅拌,据他们说Nanodrop的精度很高不用取平均值。
谢谢您的指教,我是想将来要回国教学生的时候怎么办?
这是什么意思呢?
术野有专攻,大牛也有很多弱的地方,专业的地方还是去专业的地方好。那是一个专业论坛,用google直接查丁香园就可以了。
培养了几天,浓度乃至菌的体积都可能有影响,最后溶解的TE的量,等等。
不做酶切的化,你怎么确定质粒为你所要的东西呢,切开的和不切开的跑得不一样啊。
它的大小是与质粒图一致的,不过切开看看还是有必要的,谢谢
不切是不能说“大小是与质粒图一致”。
当然如果发生位点变异也是看不出来的。
If this is your first time do this, better do it in a quantitative way. For example, you can measure OD600 to quantify the amount of E coli before extract the plasmid.
This is not quite safe since you may have other plasmid that using the same antibiotic selection. Better do a double digestion to confirm it.
Do not trust the instrument blindly. As a matter of fact, the smaller the sample size, the larger the experimental error it tend to be. Since you are not limited by the sample amount, I would suggest to measure the UV absorption on a regular UV-Vis spectrometer with proper dilution - get absorption at 260-280 nm in between 0.1-0.8 absorbance on most instruments. Don't forget the blank, it is also very important. I would use the same buffer you used to disolve plasmid in the same cuvette to measure blank. Subtract this blank from the spectrum you measured for your plasmid solution.
I did not do molecular cloning for a long time. But 260/280=1.91 is off a little bit for double strand DNA in my memory (It shows the purity of your DNA sample).
Plasmid can adopt perfect super-coil structure, it can be also nicked so that it will adopt open circle or even linear conformation. Different conformation of plasmid has different mobility rate. This is why you need to digest it in order to compare the size.
今后还要多多请教。
偶就是半瓶子醋咣当,呵呵。
我的经验是如果你检测的方法出现问题,几次的结果应该相近,如果有较大的变化,那你的手法有问题。